BoCaTFBS
L.Y.Wang, M. Snyder, M. Gerstein
In order to understand the molecular mechanisms of gene regulation, a robust method is required to discriminate transcription factor binding sites from non-binding sites on a genomic scale. Experimental methods such as ChIP-chip experiments (microarray-based readout of chromatin immuno-precipitation assays), though gaining great success, remain time-consuming, expensive, and noisy. Traditional computational methods for binding site identification, such as consensus sequences, profile methods, and HMMs, are known to generate high false positive rates when applied genome-wide. They are based on training only with positive data, the small numbers of known binding sites. Thus, we are motivated to propose a new computational method to discover transcription-factor binding sites that synthesizes the noisy data from ChIP-chip experiments with known positive binding-site patterns. Our method (which we call BoCaTFBS) uses a boosted cascade of classifiers, where each component is an individual alternating decision tree (i.e. an ADTBoost classifier). It uses known motifs, taking advantage of the inter-positional correlations within the motifs, and it explicitly integrates the massive amount of negative data from ChIP-chip experiments. We tune BoCaTFBS to reduce the false positive rate when applied genome-wide and use the cascade for optimum computational efficiency, an important consideration for genome-scale applications. We show that BoCaTFBS outperforms many traditional binding-site identification methods (such as profiles) in terms of sensitivity and specificity. We also show how its improvement is directly tied to the inclusion of the negative information from ChIP-chip experiments. Moreover, we show that BoCaTFBS can be successfully applied in the ongoing Encyclopedia Of DNA Elements (ENCODE) project, which aims to identify all functional elements in the human genome sequence.
Reference: L.Y.Wang, M. Snyder, M. Gerstein BoCaTFBS: a Boosted-cascade Learner to Refine the Binding Sites Suggested by ChIP-chip experiments, accepted by journal Genome Biology
Algorithm
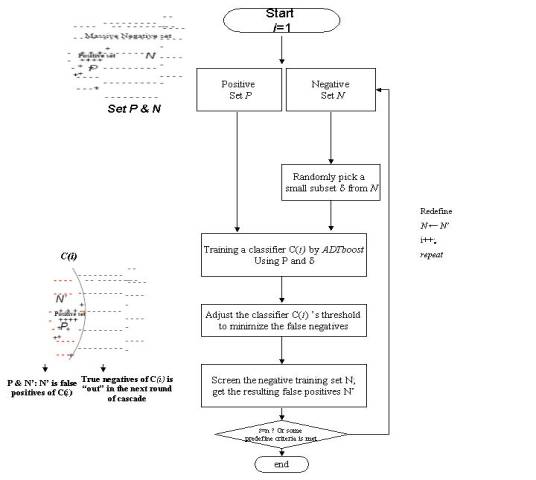
(a)
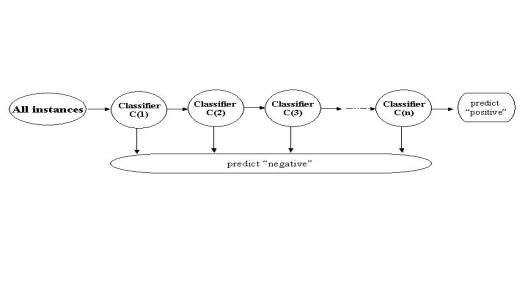
(b)
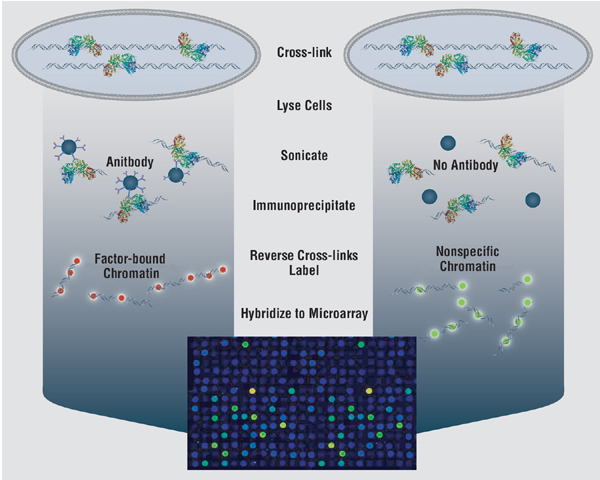
(Martone, et al)
contact: Luyong Wang